CDG: ALG1-CDG 

Inleiding
ALG1-CDG is een ernstige aangeboren stoornis van de N-glycosylatie. De ziekte wordt gekenmerkt door ernstige ontwikkelings- en psycho motorische achterstand, verlaagde spierspanning, epileptische insulten en een kleine schedel. De bloedstolling kan verstoord zijn waardoor er een hoge kans is op bloedingen of trombose, nieraandoening, ophoping van vocht in de buikholte, vergrote lever, ziekte van de hartspier, oogafwijkingen en immuundeficiëntie. De ziekte wordt veroorzaakt door mutaties in het gen ALG1 (16p13.3).
Glycoproteinen
Glycosylering is een proces in de cel waarbij suikerketens, glycanen, worden gevormd en vervolgens aan een eiwit worden verbonden. De stoffen die zo ontstaan zijn glycoproteinen. Glycoproteïnen hebben verschillende functies in het lichaam. Sommige vervullen de functie van hormonen, andere zijn nodig voor de bloedstolling, het afweersysteem of voor transporten.
Glycosylering gebeurt op speciale plaatsen binnen de cel. Het grootste deel van het glycosyleringsproces speelt zich af in het Endoplasmatisch Reticulum en in het Golgi-apparaat. Dat zijn kleine onderdeeltjes van de cel, waarin eiwitten als het ware worden ‘aangekleed’: stap voor stap worden er suikerketens opgebouwd en aan het eiwit geplakt. Er zijn meer dan honderd verschillende enzymen bij de vorming van glycoproteïnen betrokken. Als door een aangeboren afwijking één van die enzymen niet werkt of niet aanwezig is in het lichaam van een patiënt, kunnen de glycoproteïnen niet op de juiste manier gemaakt worden. Er worden dan bijvoorbeeld veel te weinig suikerketens aan het glycoproteïne geplakt. Daardoor kunnen de glycoproteïnen hun functie in het lichaam niet goed uitvoeren. Bij zulke defecten spreken we van CDG.
Omdat glycoproteïnen op zoveel plaatsen in het lichaam een belangrijke functie hebben, zorgt een fout in de productie van glycoproteïnen ervoor dat veel verschillende organen op de een of andere manier problemen hebben. CDG is dan ook een ziekte die in de meeste gevallen invloed heeft op het hele lichaam.
Veel verschillende vormen van CDG
Tot dusver zijn zo’n 100 verschillende varianten van deze ziekte geïdentificeerd waarvan de meeste zeer zeldzaam zijn. CDG-1k valt er ook onder.
Symptomen
Kinderen met ALG1-CDG hebben vaak al vanaf hun geboorte enkele opvallende afwijkingen. Ze zijn vaak heel klein bij de geboorte. Enkele patientjes hebben extra vocht en orgaanoedeem (hydrops). In heel zeldzame gevallen komt ook een multisysteemaandoening voor met grote lever en hartfunctiestoornis.
Sommige kinderen zijn geboren met gewrichtcontracturen. Andere zijn heel erg slap (hypotoon). De meeste kinderen met CDG 1k hebben typische gelaatskenmerken. Ze hebben een kleine hoofd (microcefalie), kijken scheel en kunnen heel ernstige oogafwijkingen hebben, met verlies van het gezichtsvermogen vlak na de geboorte.
Kinderen met ALG1-CDG worden geboren met een hersenafwijking die ervoor zorgt dat ze heel vaak ernstige epilepsie hebben en ook vaak meer moeite hebben met het coördineren van hun bewegingen vanwege de slapte in de spieren (hypotonie). Veel patiënten hebben daardoor moeite om te leren staan en lopen en vaak zijn ze gebonden aan een rolstoel.
Daarnaast kan microcefalie voorkomen. Dit is een aandoening van het centrale zenuwstelsel waardoor de omvang van de schedel te klein is. Dit leidt ertoe dat de hersenen zich niet volledig kunnen ontwikkelen. Ook kan de onderkaak minder gegroeid zijn waardoor de kin minder ontwikkeld is. Ook kunnen patiënten last hebben van epileptische aanvallen en mentale achteruitgang. In een deel van de kinderen is het gehoor (centraal gehoor) beperkt.
Jonge patiëntjes eten en groeien vaak slecht, waardoor het nodig kan zijn om de kinderen met een sonde (bij) te voeden. De voedingsproblemen leiden samen met de neurologische problemen in veel gevallen tot een ontwikkelingsachterstand. Dit uit zich bijvoorbeeld in spraakproblemen. Veel van de patiënten kunnen niet of nauwelijks praten, maar vaak begrijpen ze wel veel. Een spraakcomputer biedt voor zulke patiënten vaak een mogelijkheid om toch met hun ouders en omgeving te kunnen communiceren. De ontwikkelingsachterstand is ook zichtbaar in de verkrampte houding van de patiënt. Vergroeiingen kunnen erger worden als die er op jonge leeftijd ook al waren.
Er zijn verschillende laboratoriumafwijkingen, inclusief stollingsproblemen en neiging tot bloeding. Kinderen hebben ook een verhoogd risico op trombose. Ze krijgen vaak infecties. De leverfunctie kan ook afwijkend zijn.
De eerste levensjaren zijn kritiek, waarbij er ernstige infecties of problemen met de lever of het hart op kunnen treden. Het gevolg hiervan kan zijn dat het patiëntje al op jonge leeftijd komt te overlijden.
Sinds kort weten we dat patiënten volwassen kunnen worden en de diagnose pas krijgen na de leeftijd van 20 jaar. Deze patiënten hebben een mildere aandoening met epilepsie, een ontwikkelingsachterstand en vaak ook een taalachterstand.
Diagnose
Er is een laboratoriumtest waarmee patiënten kunnen worden gescreend op CDG-defecten. In die test wordt het bloed onderzocht op transferrine, een glycoproteïne. De test laat zien of het transferrine normaal gevormd is, of dat er afwijkingen zijn in de suikerketens van het transferrine. Als het transferrine afwijkend is, is dat een aanwijzing dat de patiënt één van de varianten van CDG heeft. Uit het testresultaat is echter niet af te lezen welke variant de patiënt heeft. Bovendien kan de transferrinetest ook een afwijkende uitslag geven bij sommige andere stofwisselingsziekten. Daarom is het vrijwel altijd nodig om vervolgonderzoeken te doen. Daarvoor wordt vaak extra bloed afgenomen, of een stukje huid.
Met de extra laboratoriumonderzoeken is het voor artsen mogelijk om het precieze enzymdefect aan te wijzen dat de ziekte van de patiënt veroorzaakt. Dat is niet alleen nodig om de diagnose met zekerheid te kunnen stellen, maar is ook belangrijk als de ouders van de patiënt graag nog meer kinderen willen krijgen.
Wanneer screenen op CDG?
Patiënten met CDG hebben soms zulke uiteenlopende symptomen, dat het moeilijk is om te bepalen of een transferrinetest zinvol is. Over het algemeen zullen artsen een transferrinetest doen als de patiënt een onverklaarbare verstandelijke of motorische ontwikkelingsachterstand heeft, in combinatie met één of meerdere orgaanproblemen.
Omdat CDG erg zeldzaam is, kan het soms moeilijk zijn om de juiste diagnose te vinden. Toch zijn er steeds meer artsen die wel eens van deze groep van ziekten gehoord hebben. Daardoor zijn er in de laatste jaren veel meer patiënten gediagnosticeerd en zijn er verschillende nieuwe varianten van de ziekte ontdekt.
Prenatale diagnostiek
Omdat er zoveel verschillende soorten CDG zijn, is prenatale diagnostiek alléén mogelijk als het precieze enzymdefect of genetisch defect van een patiënt bekend is. Alleen dan kunnen ouders bij een volgende zwangerschap laten testen of het ongeboren kindje de ziekte ook heeft.
Behandeling
ALG1-CDG is niet te genezen. Het is alleen mogelijk om met verschillende behandelingen de symptomen van de ziekte zoveel mogelijk te verlichten. Logopedie en veel stimulatie zijn essentieel in de behandeling. Zo kan een kind een neusmaagsonde krijgen als het veel moeite heeft met eten. Ook wordt vaak uit voorzorg antibiotica gegeven om infecties te voorkomen. Het is van belang dat de patiëntjes regelmatig onder controle blijven van een kinderarts metabole ziekten, zodat eventuele problemen vroegtijdig kunnen worden ontdekt.
Erfelijkheid
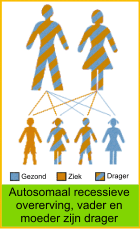 Stofwisselingsziekten zijn erfelijke ziekten. Dat betekent dat je met de ziekte geboren wordt en er niet van kan genezen. Het betekent meestal dat de ouders van te voren niet hadden kunnen weten dat hun kind ziek zou worden. In de meeste gevallen hebben beide ouders nergens last van. Zij zijn dan ‘gezonde dragers’ van een afwijkend gen en hebben daarnaast het normale gen (zie onder). Het normale gen zorgt ervoor dat het benodigde enzym bij hen voldoende aangemaakt wordt. Een kind met de ziekte heeft twee afwijkende genen en mist het normale gen. Daardoor wordt het enzym niet of onvoldoende aangemaakt.
Stofwisselingsziekten zijn erfelijke ziekten. Dat betekent dat je met de ziekte geboren wordt en er niet van kan genezen. Het betekent meestal dat de ouders van te voren niet hadden kunnen weten dat hun kind ziek zou worden. In de meeste gevallen hebben beide ouders nergens last van. Zij zijn dan ‘gezonde dragers’ van een afwijkend gen en hebben daarnaast het normale gen (zie onder). Het normale gen zorgt ervoor dat het benodigde enzym bij hen voldoende aangemaakt wordt. Een kind met de ziekte heeft twee afwijkende genen en mist het normale gen. Daardoor wordt het enzym niet of onvoldoende aangemaakt.
Autosomaal recessief
In elke cel van het lichaam is het erfelijke materiaal in tweevoud aanwezig en is verdeeld in chromosoomparen. Er zijn per cel 22 gelijke paren (autosomen), terwijl het 23e paar geslachtsbepalend is en verschilt tussen een vrouw, die twee X-chromosomen heeft en een man die een X- en een Y-chromosoom heeft.
Deze Stofwisselingsziekte erft ‘autosomaal recessief’ over. Autosomaal betekent dat het afwijkende gen op één van de 22 gewone chromosomen ligt. Zowel jongens als meisjes kunnen ziek zijn. Daarnaast is een afwijkend gen op een van de twee chromosomen ondergeschikt aan het normale gen op het andere chromosoom (recessief), die in dat geval compenseert. Dit gebeurt bij een “gezonde dragers”, die de ziekte dus niet zal krijgen. Er zijn dus twee afwijkende genen nodig om de ziekte te krijgen. Een kind met een stofwisselingsziekte heeft van allebei zijn ouders een afwijkend gen geërfd.
De ouders zijn niet ziek, maar zijn wel drager van het afwijkende gen. Daardoor hebben ze 25% kans (1 op 4) bij elke zwangerschap op een kind met de ziekte. Ook hebben ze 75% (3 op 4) kans op een kind dat niet ziek is. Daarvan zal 2/3, net als de ouders, gezonde drager zijn. Deze kinderen kunnen de ziekte alleen doorgeven als hun partner ook dezelfde afwijking heeft op zijn DNA.
Overige informatie
Omim nummer
Synoniemen:
Mannosyltransferase 1-deficiëntie
Congenitale defecten van de glycosylering type 1k
Congenital disorder of glycosylation, type 1k
CDG1K
Koolhydraatdeficiënt glycoproteïne-syndroom type Ik
Meest gebruikte naamCDG- ALG1-CDG
Kenniskaarten:
Informatie voor kinderen:
Zijn er leden met deze ziekte?
Er is één lid met ‘CDG: ALG1-CDG’ bij ons aangemeld.
Datum laatst bewerkt:
02 August 2021
Autorisatie door:
E. Morava
Disclaimer
Aan de ziekte-informatie kunnen geen rechten worden ontleend. De informatie is mogelijk niet op alle punten actueel, omdat de ontwikkelingen en inzichten snel kunnen gaan. VKS tracht de ziekte-informatie zo goed mogelijk actueel te houden.
Ervaringsverhalen zijn persoonlijke verhalen. De beschrijving van de ziekte en symptomen gelden voor deze persoon. Zoals voor veel erfelijke ziekten geldt, is er een behoorlijke variatie in ernst onder de patiënten. U kunt uit dit verhaal dan ook geen algemene conclusies trekken. Het verhaal geeft slechts een beeld hoe het leven met deze stofwisselingsziekte in de praktijk eruit kan zien.
Heeft u hulp nodig bij het inloggen?
Vond u deze informatie nuttig? Help ons dan om dit in stand te houden.
Een groot aantal van de lemma's in deze verklarende woordenlijst is samengesteld door Barbara Trimbos-Hart, eigenaar/beheerder van http://www.mitoinfo.nl, site voor en door volwassenen met een mitochondriële aandoening.
Wilt u zelf een woord toevoegen? Mail dan term en omschrijving naar info@stofwisselingsziekten.nl
A- voor een woord betekent niet aanwezig, bijvoorbeeld a-typisch.
Aanpassingen Dagelijks Leven, zelfredzaamheid. Toepassingen van functies en vaardigheden bij bijvoorbeeld: voortbewegen, eten en drinken, wassen, aan- en uitkleden, toiletgebruik etc.
Vanaf de geboorte aanwezig (congenitaal). Aangeboren aandoeningen zijn afwijkingen die ontstaan in de baarmoeder en waarmee het kind geboren wordt. Ze kunnen erfelijk zijn, maar ze kunnen ook ontstaan door invloeden van buitenaf, bijvoorbeeld door medicijn- of alcoholgebruik tijdens de zwangerschap, of door besmetting van de moeder met bijv. rode hond. Ook kan er sprake zijn van een combinatie van erfelijke en omgevingsfactoren (bijv. open ruggetje (spina bifida)).
Abductie is de buitenwaartse of zijwaartse beweging van een lichaamsdeel. Adductie is het tegenovergestelde van abductie en betekent dus te dicht bij elkaar.
Kortstondige bewustzijnsdaling als gevolg van een stoornis in de hersenen
Zuurvergiftiging. Bij acidose is er een toename van de zuurgraad in het bloed. Dit geeft een daling van de pH-waarde.
Adductie is de binnenwaartse beweging van een lichaamsdeel. Bij een adductiestand van bijvoorbeeld de benen bestaat er geen ruimte tussen de benen.
Abductie is het tegenovergestelde van adductie en betekent dus te gespreid.
Vijf enzymcomplexen die ingebed liggen in de binnenmembraan van het mitochondrion.
In het proces om energie te maken worden zuurstofmoleculen verbruikt zodat er gesproken kan worden over de 'ademhaling op celniveau'. De ademhalingsketen, of oxidatieve fosforylering, is het hart van de energiecentrale van het lichaam.
Afkorting van adenosinedifosfaat. Het energiearme product dat geproduceerd wordt wanneer ATP energie in de cel gebracht heeft.
Verworven taalstoornis ten gevolge van hersenletsel. Met spreekt van afasie als de taalstoornis ontstaat nadat de taalontwikkeling is afgerond. Een aangeboren taalstoornis wordt dysfasie genoemd.
Letterlijk: lopend. Een ambulante patiënt wordt niet in het ziekenhuis opgenomen maar heeft een kortdurend bezoek, bijvoorbeeld poliklinisch.
De bouwstenen van eiwitten
Het eerste gedeelte van een medisch onderzoek waarbij inlichtingen aan / over de patiënt gevraagd worden. De anamnese komt vooral tot stand door specifieke vragen van de arts. De anamnese omvat bijvoorbeeld:
* algemene gegevens, zoals naam, geboortedatum, beroep
* de ontwikkeling van de ziekte
* vroegere ziekten
* gegevens over de familie.
Anamnestisch verkregen informatie is informatie die de arts heeft verkregen door vragen aan de patiënt (ouders).
Het eerste gedeelte van een medisch onderzoek waarbij inlichtingen aan / over de patiënt gevraagd worden. De anamnese komt vooral tot stand door specifieke vragen van de arts. De anamnese omvat bijvoorbeeld:
* algemene gegevens, zoals naam, geboortedatum, beroep
* de ontwikkeling van de ziekte
* vroegere ziekten
* gegevens over de familie.
Anamnestisch verkregen informatie is informatie die de arts heeft verkregen door vragen aan de patiënt (ouders).
Zuurstoftekort in de weefsels
Medicatie tegen epilepsie. Wordt soms ook voorgeschreven bij zenuwklachten.
Middel tegen stuipen, toevallen (bij epilepsie). Synoniem: anticonvulsant.
Stof die oxidatie, dat is binding met zuurstof, tegengaat. Bepaalde vitamines (vit. E) zijn bijvoorbeeld antioxidanten.
Kort stoppen met ademen, ademstilstand, bijvoorbeeld in de slaap (slaapapnoe)
Stoornis in de samenwerking tussen de spieren, waardoor het verrichten van ordelijke bewegingen moeilijk wordt. Er zijn coördinatiestoornissen. Atactische bewegingen zijn onregelmatige, onbeheerste bewegingen.
Onophoudende, onwillekeurige, langzame buig- en strekbewegingen van vingers en tenen
Type epileptische aanval waarbij er geen verstijving van de spieren optreedt, maar juist een verslapping, waardoor de betrokkene plotseling bewusteloos neervalt.
Afkorting van adenosinetrifosfaat. De energie die via een groot aantal stofwisselingsstappen wordt vrijgemaakt en opgeslagen opdat de cel er iets mee kan.
ATP wordt gevormd uit ADP en fosfaat; dit proces heet oxidative phosphorylation (OXPHOS).
Verschrompeling van een orgaan door het afsterven van de cellen waaruit het orgaan bestaat.
Het afsterven wordt veroorzaakt door een tekort of juist een overmaat aan stoffen die de cel nodig heeft om te blijven leven.
Alleen voor de betrokkene waarneembare (=subjectieve) ervaringen, zoals het ruiken, zien, horen, proeven of voelen van iets, als voorbode van een epileptische aanval of migraine-aanval.
Het gedeelte van het zenuwstelsel dat de onwillekeurige organen (de buiten de wil om functionerende organen) verzorgt, zoals die van de spijsvertering, de bloedsomloop en de ademhaling.
Afwijkende voetzoolreflex, waarbij de grote teen bij het strijken onder de voetzool naar boven beweegt in plaats van naar onder. Deze afwijkende voetzoolreflex wijst op een stoornis in het zenuwstelsel.
BAEP staat voor Brain stem Auditory Evoked Potential. Dit is een test waarbij de zenuwgeleiding tussen oor en hersenen wordt onderzocht. Met een BAEP-test wordt de reactie van de hersenen gemeten op een prikkel van het gehoorsysteem. Doel is informatie te verkrijgen over de werking van de gehoorzenuw. Het onderzoek is niet pijnlijk en de patiënt hoeft zelf niets te doen.
Bij metabole ziekten is dat meestal de aanduiding voor onderzoek in bloed of urine, waarbij gekeken wordt naar afwijkende metabolieten (zie aldaar). Een afwijkende hoeveelheid van bepaalde stoffen kan aanleiding zijn voor verder gericht onderzoek, door middel van enzymdiagnostiek (zie aldaar) of gericht onderzoek in het DNA.
Het voorvoegsel brady- betekent langzaam of traag, bijvoorbeeld bradycardie: trage hartslag, bradyfrenie: mentale traagheid, vertraagd denken.
Een arts die gespecialiseerd is in het opsporen, diagnosticeren en behandelen van ziekten van het hart.
Aandoening van de hartspier
Carnitine (carnitene) is een aminozuur dat normaal in het eten voorkomt en door het lichaam gebruikt wordt voor het leveren van energie aan de skeletspieren. Carnitine is nodig voor het transport van vetzuren naar de mitochondriën zodat het vet kan worden verbrand. Daarnaast zou carnitine ook de membraan (wand) van de mitochondriën sterker maken.
Microscopisch klein onderdeel van alle planten, dieren en mensen. Het menselijk lichaam bestaat uit miljarden cellen.
Het centrale onderdeel van de cel waar het erfelijk materiaal, het DNA, in opgesloten zit. Vanuit de celkern vindt de aansturing van alle processen in de cel plaats.
De kleine hersenen betreffend
Een onderdeel van de hersenen, ook bekend als de kleine hersenen. De kleine hersenen hebben een belangrijke functie bij het sturen en regelen van bewegingen. De uiterlijk waarneembare verschijnselen van het niet goed functioneren van de kleine hersenen wordt ataxie (zie daar) genoemd.
Choreoathetose is een extrapiramidale stoornis. Mensen met deze bewegingsstoornis hebben een combinatie van twee bewegingsstoornissen: chorea en athetose. Chorea wordt gekenmerkt door onwillekeurige, schokkende, snelle bewegingen. Athetose wordt gekenmerkt door onwillekeurige, kronkelende bewegingen die langzamer zijn.
Drager van erfelijke eigenschappen. Het belangrijkste component is DNA. Alle chromosomen zitten samen in de celkern.
CK=creatine kinase. Door extreme spierarbeid kan er, door beschadiging van het vliesje om de spier heen, lekkage van weefselvocht uit de spieren ontstaan. Om de mate van lekkage uit spiervezels te meten kan de CK-waarde in het bloed gemeten worden. De CK-activiteit is bij veel, maar niet alle, spierziekten verhoogd. Zeer hoge waarden vindt men bijvoorbeeld bij spierdystrofie, normale waarden bij sommige aangeboren spierziekten. Als bij laboratoriumonderzoek een verhoogde CK-activiteit wordt gevonden, dan zal de bepaling in de regel herhaald worden om te kijken of de verhoging tijdelijk is en veroorzaakt wordt door een ongewoon sterkte inspanning.
Een abnormale beweging die gekarakteriseerd wordt door snelle aan- en ontspanning van spieren.
Een abnormale beweging die gekarakteriseerd wordt door snelle aan- en ontspanning van spieren.
Afkorting van Central Nervous System, de Engelstalige term voor het centraal zenuwstelsel.
Het denkvermogen. Het vermogen om alle informatie uit het dagelijks leven te interpreteren:
* oriëntatie in tijd, plaats en persoon
* aandacht en concentratie
* tempo van informatieverwerking
* geheugen
* schoolse vaardigheden
* redeneervermogen
* leervermogen
Met betrekking tot het denkproces
Aangeboren, reeds vanaf de geboorte aanwezig
Het in een gedwongen stand staan van een gewricht. Noch de patiënt zelf noch een ander kan die stand veranderen. Een contractuur kan ontstaan doordat de 'strekkers' ziek zijn en de 'buigers' gaan overheersen.
Een MRI of CT-scan 'met contrast' betekent dat er een contrastvloeistof ingespoten wordt.
Afkorting voor het Centraal ZenuwStelsel (hersenen en ruggenmerg)
Vermindering, afwezig zijn, niet functionerend
Benaming voor allerlei veranderingen in cellen en weefsels waardoor hun normale functie aangetast wordt. Een langzaam, geleidelijk proces van afsterven van bepaalde groepen (hersen)cellen.
Zie ook Atrofie.
Uitdroging, overmatig verlies van lichaamsvocht
Verslechteren, achteruitgaan
Dys- voor een woord betekent afwijkend.
Dysartrie is een spraakstoornis. De spieren die nodig zijn voor de stemgeving, het ademen en de uitspraak werken door een beschadiging van het zenuwstelsel onvoldoende. Omdat mensen met dysartrie moeilijk te verstaan zijn kan de communicatie problemen opleveren door bijvoorbeeld onduidelijke uitspraak, nasaal (door de neus) spreken of een combinatie hiervan.
Stoornis in het vermogen om woorden en zinnen te vormen, zonder dat het denkvermogen is aangetast.
Stoornis in de spierspanning door een fout in de coördinatie van de hersenen bij het bewegen. Hierdoor ontstaan continue samentrekkingen van de spieren waar de patiënt zelf geen invloed op kan uitoefenen.
Electro Cardiogram. Onderzoek naar de hart-activiteit. Er worden hiervoor plakkertjes op de borst geplaatst. Het onderzoek is pijnloos en kortdurend.
Electromyogram is een spier/zenuw onderzoek. Soms volledig pijnloos, soms onaangenaam door de schokjes en het plaatsen van kleine naaldjes.
Elektro-encefalogram. Registratie van de elektrische activiteit in de hersenen. Met lijm of een soort badmuts worden elektroden op het hoofd geplakt. Behalve ogen sluiten en openen hoeft de patiënt niets te doen. Het is een pijnloos onderzoek.
Huidverdovingsmiddel in de vorm van crème of pleister dat gebruikt kan worden als verdoving bij een injectie, plaatsen infuus, biopt of lumbaalpunctie.
Niet gedefinieerde ziekte in de hersenen.
Aandoening van de hersenen en de spieren.
Eiwit dat in ons lichaam wordt gemaakt door bepaalde aminozuren aan elkaar te knopen op basis van genetische informatie uit de celkernen.
Voor de diagnose van een stofwisselingsziekte is onderzoek in bloed of lichaamsweefsel noodzakelijk. Meestal wordt voor dat laatste een huidbiopt gebruikt, bij sommige aandoeningen, spier- of leverweefsel. Bij het enzym onderzoek worden de activiteiten van de verschillende enzymen op celniveau gemeten in een gespecialiseerd laboratorium. Enzymdiagnostiek komt in beeld als de behandelend arts bij een patient aanwijzingen vindt voor één of meer erfelijke metabole ziekte(n) met een bekend enzymdefect.
Een paramedische discipline die zich bezig houdt met onderzoek, begeleiding en behandeling van zo zelfstandig mogelijk functioneren.
* zelfverzorging, ADL activiteiten (zie aldaar), eten, drinken, wassen, aankleden, naar toilet gaan
* huishouden
* verplaatsen en vervoer
* vrije tijd / hobby's
* werk
* wonen
Meestal plotseling optredende verslechtering. Ook wel 'crash' genoemd.
Bij extrapiramidale bewegingsstoornissen is de aansturing van de spieren verstoord. Symptomen zijn bijv. spiertrekkingen in het gezicht, krampachtige strekkingen van het lichaam, trillende handen en abnormale onwillekeurige bewegingen die de normale bewegingen hinderen.
Het aangezicht betreffende.
Huidcellen. Kunnen onderzocht worden d.m.v. een huidbiopt.
De dragers van erfelijke eigenschappen die samen het DNA vormen. Genen bevatten het 'recept' voor het maken van eiwitten, waaronder enzymen.
Ander woord voor druivensuiker. De belangrijkste energiebron voor de cellen.
Glucosevoorraad in de lever.
Grote of grand mal : Een bepaald type epileptische aanval, die in twee fasen verloopt, namelijk eerst een tonische fase (waarin alle spieren secondelang verstijven en de ademhaling stokt) en vervolgens een clonische fase (waarin de spieren heftige ongecontroleerde bewegingen maken. Wordt ook wel grand mal-aanval of tonisch-clonische aanval genoemd.
Gezichtsveldverlies t.g.v. hersenletsel aan 1 of beide buitenkanten van het gezichtsveld.
Halfzijdige verlamming (arm + been + evt gezicht).
Het bloed betreffende.
Hyper- voor een woord staat voor te veel, overmatig.
Overmatige, ongewilde, ondoelmatige beweging.
Verhoogde reactie op prikkels. Te heftige reflex.
Grote afstand tussen b.v. de ogen.
Hoge bloeddruk.
Hypo- voor een woord staat voor te weinig, onvoldoende.
Hypoglykemisch, oftewel hypoglykemie, wil zeggen dat je een te laag gehalte aan bloedsuiker (glucose) hebt. Je kunt dan last hebben van de volgende verschijnselen: zweten, trillen, angst, hartkloppingen, je kunt je niet meer concentreren, verwarring, vermoeidheid, moeite met spreken, slapte, hoofdpijn, misselijkheid, wazig zien.
Hypoglykemisch, oftewel hypoglykemie, wil zeggen dat je een te laag gehalte aan bloedsuiker (glucose) hebt. Je kunt dan last hebben van de volgende verschijnselen: zweten, trillen, angst, hartkloppingen, je kunt je niet meer concentreren, verwarring, vermoeidheid, moeite met spreken, slapte, hoofdpijn, misselijkheid, wazig zien.
Lage bloeddruk.
Het steeds erger trillen van een ledemaat, naar gelang het doel dichterbij komt.
Toediening van een geneesmiddel per injectie in een spier.
Toediening van een geneesmiddel per injectie in een ader.
Dieet gekenmerkt door voeding met een hoog vetgehalte en een laag koolhydraat- en eiwitgehalte. Door dit dieet mist het lichaam de koolhydraten en moet het op zoek naar een andere energiebron: Het schakelt over op vetverbranding. Kan worden toegepast bij anders slecht behandelbare epilepsie en bij sommige stofwisselingsziekten als alternatieve energiebron.
Een arts gespecialiseerd in de erfelijkheid van ziekten.
Melkzuur.
Regelcentrum in de hersenen dat informatie vanuit al onze zintuigen krijgt en verwerkt en verder betrokken is bij geheugen, gedrag en emoties.
Het vocht in de hersenen en het ruggenmerg . Afwijkingen in het liquor (afgetapt m.b.v. LP =Lumbaalpunctie) wijzen op een encephalopathie. Het soort afwijkingen kan richting geven aan het vinden van een diagnose.
Een paramedische discipline die zich bezig houdt met het onderzoeken, begeleiden en behandelen van alles dat met communicatie te maken heeft:
* primaire mondfuncties (zie aldaar)
* spraak
* taal
* gehoor
Door middel van een prik tussen de ruggenwervels wordt vocht afgetapt dat zich in het ruggenmerg bevindt. Dit vocht, ook wel liquor genoemd, wordt onderzocht. De uitslag van dit onderzoek kan helpen een diagnose te stellen. Na de prik kun je hoofdpijn hebben of duizelig zijn. Dit gaat meestal vanzelf weer over. Plat blijven liggen en veel drinken helpt. Als er na 5 dagen nog hoofdpijn bestaat contact opnemen met de arts.
De tussen- of eindproducten die ontstaan nadat een chemische stof in een biologisch systeem (bacteriën, planten ,dieren en mensen) metabolisme heeft ondergaan. Metabolieten zijn onder andere : de aminozuren, adenosinetrifosfaat of ATP, glucose, adrenaline enz.
Stofwisseling.
Alle biochemische reacties die plaats vinden in het lichaam. Al deze reacties samen zorgen voor de energie die het lichaam nodig heeft voor de lichaamsfuncties zoals ademhaling, denken en bewegen.
Stofwisselings (metabole) ziekte. Veroorzaakt door een fout in 1 van de vele stappen (reacties) in de stofwisseling. In de mitochondria vinden meer dan 100 verschillende stofwisselingsstappen plaats.
Een heel belangrijke functie van de mitochondria is de rol als energie-centrale in de cel. Hoewel de mitochondria veel meer functies hebben wordt met een mitochondriële aandoening vrijwel altijd een energie-ziekte bedoeld.
Mitochondriële aandoeningen kunnen op verschillende manieren ingedeeld worden.
* Naar het complex dat niet goed werkt (deficiënt is):
Complex 1, 2, 3, 4, 5 deficiëntie. Sommige deficiënties komen vaker voor dan andere (complex 1 en 4). Vaak komen er ook meer deficiënties tegelijk voor ( bijv. complex 1 + 4)
* Naar de aard van de klachten, in een syndroom (bijv. Leigh-, MERFF-, MELAS-, Kearns-Sayre-, LHON- Pearsons's en NARP syndroom).
Stofwisselings (metabole) ziekte. Veroorzaakt door een fout in 1 van de vele stappen (reacties) in de stofwisseling. In de mitochondria vinden meer dan 100 verschillende stofwisselingsstappen plaats.
Een heel belangrijke functie van de mitochondria is de rol als energie-centrale in de cel. Hoewel de mitochondria veel meer functies hebben wordt met een mitochondriële aandoening vrijwel altijd een energie-ziekte bedoeld.
Mitochondriële aandoeningen kunnen op verschillende manieren ingedeeld worden.
* Naar het complex dat niet goed werkt (deficiënt is):
Complex 1, 2, 3, 4, 5 deficiëntie. Sommige deficiënties komen vaker voor dan andere (complex 1 en 4). Vaak komen er ook meer deficiënties tegelijk voor ( bijv. complex 1 + 4)
* Naar de aard van de klachten, in een syndroom (bijv. Leigh-, MERFF-, MELAS-, Kearns-Sayre-, LHON- Pearsons's en NARP syndroom).
Het gedeelte van de cel dat verantwoordelijk is voor de omzetting van allerlei stoffen in energie. Mitochondriën worden ook wel de energiecentrales van het lichaam genoemd.
Meervoud: Mitochondriën of mitochondria.
Beweging. Wat met bewegen te maken heeft (zitten, staan, lopen,… schrijven, tekenen,…).
Beweging. Wat met bewegen te maken heeft (zitten, staan, lopen,… schrijven, tekenen,…).
Iemand met een genetische aandoening kan zowel normale als abnormale cellen hebben. Dan zit de fout dus niet in alle cellen. De klachten van de ziekte zijn dan minder of minder erg of kunnen zelfs ontbreken. Dit noemen we mozaïekvorm of mozaïcisme.
Iemand met een genetische aandoening kan zowel normale als abnormale cellen hebben. Dan zit de fout dus niet in alle cellen. De klachten van de ziekte zijn dan minder of minder erg of kunnen zelfs ontbreken. Dit noemen we mozaïekvorm of mozaïcisme.
Magnetic Resonance Imaging. Dit onderzoek is te vergelijken met de CT-scan. Je ligt hierbij in een tunnel. Er worden opnamen gemaakt van de hersenen of het ruggenmerg. Het apparaat, waar je met je hoofd inligt, kan deze opnamen maken door middel van het veranderen van magnetische velden. Er worden geen röntgenstralen maar radiogolven gebruikt. De opnamen van hersenen en ruggenmerg zijn van uitzonderlijk goede kwaliteit. Omdat er gebruik gemaakt wordt van magnetische golven mag je geen voorwerpen van ijzer dragen of andere voorwerpen die het magnetisch veld kunnen verstoren. Tijdens het onderzoek is een hard getik hoorbaar. Je kunt oordoppen dragen of een bandje met favoriete muziek meenemen en beluisteren. Bij kinderen wordt veelal een lichte narcose gegeven omdat het onderzoek langdurig stilliggen vereist. Soms wordt contrastvloeistof ingespoten. Het onderzoek is ongevaarlijk en een belangrijk middel om een diagnose te kunnen stellen. Idem: NMR, Nuclear Magnetic Resonance
Een onderzoeks- of behandelteam dat bestaat uit meerdere specialisten / specialisaties die samen werken. Een revalidatie-team is multidisciplinair. Naast de revalidatiearts kunnen o.a. de fysiotherapie, ergotherapie,logopedie maatschappelijk werk, psychologie, diëtist, verpleegkundige deel uit maken van een dergelijk team.
Spier.
Een musculaire aandoening is een aandoening van de spieren (myopathie).
Een verandering in het genetisch materiaal. Zowel in het DNA als in de genen.
Spierpijn.
Witte stof.
Vetachtige cellen die een isolatielaag vormen rondom zenuwcellen. Deze laag maakt het mogelijk de elektrische signalen die de cel verzendt, sneller te vervoeren. Bij de geboorte is myeline nauwelijks aanwezig. In de ontwikkeling naar volwassenen wordt het stapsgewijs aangemaakt.
Myo- (grieks) is spier.
Wanneer en voor een woord MYO staat, dan betekent het dat het de spieren betreft.
Willekeurige, snelle spierschokken met kortdurend bewegingseffect. Niet alle myoclonieën zijn epileptisch en ze kunnen bij vele aandoeningen voorkomen.
Type epileptische aanval waarbij enkelvoudige of in reeksen voorkomende spierschokken optreden in de armen en/of de benen, met een zeer kortdurende bewustzijnsstoornis. Het kan een voorbode zijn van een volledige epileptische aanval (zie Tonisch-clonische aanval).
Iedere abnormale conditie of ziekte van het spierweefsel. Kan zowel de skeletspieren als hartspier betreffen.
Bij myotonie ontspannen aangespannen spieren te langzaam ten gevolge van problemen in de spier- of zenuwcellen. Iemand met myotonie heeft bijvoorbeeld moeite met het loslaten van een voorwerp dat hij eerder vast had. Het gevoel wordt wel omschreven als 'stijfheid'.
Het afsterven van zenuwcellen in het netvlies, waardoor het gezichtsvermogen slechter wordt: Men ziet waziger, niet meer geheel scherp.
Zenuwpijn.
Zenuwontsteking.
Met betrekking tot de zenuwen en / of hersenen.
De zenuwen en spieren betreffende.
Ziekte van de zenuwen.
Een verzameling van uiteenlopende stoffen die zorg dragen voor en/of informatie doorgeven van de ene zenuwcel naar de andere of van zenuwcellen naar klieren, spieren, zintuigcellen en terug.
met betrekking tot de hersenen, het ruggemerg en een onderdeel van het zenuwstelsel dat zelfstandig (buiten de wil om) zorgt voor het aansturen van verschillende organen
met betrekking tot de hersenen, het ruggemerg en een onderdeel van het zenuwstelsel dat zelfstandig (buiten de wil om) zorgt voor het aansturen van verschillende organen
Nuclear Magnetic Resonance. Zie MRI
Het onderzoek waarbij oogbewegingen worden geregistreerd.
Onwillekeurige, snelle, ritmische bewegingen (trillen) van het oog, meestal horizontaal. Kan aanwijzing zijn voor aandoening in evenwichtsorgaan.
Registratie van de oogbewegingen.
Zie ook nystagmografie.
Oogspierverlamming.
Onderdelen van de cel. De celkern is een van de organellen. Daarnaast zijn bekend: lysosomen, peroxisomen, mitochondriën, het Golgiapparaat enzovoort.
Het gehoor, het oor betreffende.
Oxidatieve Phosphorylering.
De omzetting van energie verkregen uit de verbranding van suikers en vetten tot lichaamseigen energie ter hoogte van de ademhalingsketen in de celkern.
Niet pijnlijke gevoelens die niet worden veroorzaakt door een passende uitwendige prikkel. Ook wel sensibiliteitsstoornissen genoemd. Bijvoorbeeld prikkelen, tintelen of een bandengevoel.
Volledig krachtsverlies.
Verlamming aan beide benen.
Krachtsvermindering. Aanduiding van de mate van verlamming.
Pathie (grieks) betekent ziekte, afwijking, aandoening. Pathie komt achter datgene dat afwijkend is. Myo-pathie betekent een aandoening van de spieren. Neuro-pathie betekent een aandoening van de zenuwen. Pathologisch is door een ziekte veroorzaakt.
Door een ziekte veroorzaakt.
Gezichtsveld-onderzoek.
Persoonlijkheid is het geheel van kenmerken van iemands gedrag en heeft altijd een relatie met de buitenwereld. Elementen die samen de persoonlijkheid vormen:* basisstemming
* zelfbeeld en vertrouwen
* impulscontrole
* evaluatie van eigen gedrag
* inzicht in en acceptatie van stoornissen en mogelijkheden
* oordeelsvermogen
* sociaal gedrag
* persoonlijkheidsstoornissen.
Positron Emissie Tomografie. Techniek om een beeld van de hersenen krijgen. Door een combinatie van twee methoden geeft de functie onder andere bloeddoorstroming, zuurstof- en glucoseverbruik in de hersenen en de werking van de hersenen weer, wordt ook gebruikt om epileptische haarden op te sporen.
Voor de geboorte.
Specialisatie binnen de logopedie die zich bezig houdt met mondfuncties voordat de spraak / taal op gang gekomen is (bijvoorbeeld zuigen, slikken, kauwen).
Voorspelling omtrent het verdere verloop.
Toenemend in ernst.
Psychische gevolgen van een ziekte kunnen de
* cognitie
* intelligentie
* persoonlijkheid en gedrag betreffen.
Intelligentie = kennis- en begripsvermogen. Cognitie = denkvermogen.
Intelligentie en cognitie zijn noodzakelijk om te kunnen leren en vormen de basis voor adequaat gedrag.
Hangend ooglid.
Druivenzuur.
Verlamming aan benen en armen.
Geestelijke achterstand.
Netvlies. Het vlies dat de binnenvlakte van het oog bekleedt en waarop het beeld van de waargenomen voorwerpen wordt gevormd. Netvliesdegeneratie: Het afsterven van zenuwcellen in het netvlies, waardoor het gezichtsvermogen slechter wordt: Men ziet waziger, niet meer geheel scherp.
Erfelijke aandoening van het netvlies (ogen).
Achter.
Behandelonderdeel van (onder andere) neuromusculaire aandoeningen. Bij progressieve achteruitgaande)aandoeningen gericht op voorkoming van het verlies van onafhankelijkheid en kwaliteit van bestaan.
De behandelaars die betrokken kunnen zijn bij de revalidatie. Revalidatie-arts, stelt een revalidatie-diagnose en een revalidatie-plan -doel. Coördineert de verschillende therapieën en andere bij de patient betrokken specialisten(case-management). Verder kunnen van het team deel uit maken: De fysiotherapeut, ergotherapeut, logopedist, maatschappelijk werker, psycholoog, orthopedagoog, orthopedisch schoen- en/of instrumentenmaker, diëtist, verpleegkundige, muziektherapeut. De huisarts maakt geen deel uit van het revalidatieteam, maar dient wel op de hoogte gehouden worden door de revalidatiearts. De revalidatiearts dient het aanspreekpunt voor de huisarts te zijn.
Verkromming van de wervelkolom.
Kalmerend, rustgevend medicijn.
Met het lichaam te maken hebbende. Somatische gevolgen van een ziekte kunnen
* motorisch,
* vegetatief,
* neurologisch,
* orthopedisch en
* sensorisch zijn.
Motorisch = beweging; vegetatief = basale lichaamsprocessen; neurologisch = zenuwstelsel; orthopedisch = botten en gewrichten; sensorisch = waarneming.
Verlamming die gepaard gaat met een verhoogde spierspanning (tonus, hypertonie).
Somato-sensory evoked potentials. Bij dit onderzoek worden de zenuwbanen onderzocht die het gevoel voorgeleiden. Hiervoor worden elektrische prikkels op scheenbeen of pols toegediend.
Processen in het lichaam waarbij in de cel de ene stof wordt omgezet in de andere.
Ook: metabole ziekte. Door gebrek aan een bepaald enzym of activator van een enzym treedt een stoornis in het geheel van biochemische reacties die het lichaam aansturen. De plaats, aanwezigheid van restactiviteit, en de functie van het deficiënte enzym bepalen de gevolgen voor het lichaam en dus de symptomen van de ziekte die ontstaat.
Scheelzien.
Een combinatie van verschillende klachten / symptomen die gezamenlijk een ziektebeeld vormen.
Transcutane Electro Neuro Stimulatie. Een klein toestelletje (krachtbron) dat door middel van electroden en klevers wordt verbonden met de pijn-zone. De pijngewaarwording wordt verminderd door elektrische stimulatie (tinteling). Het TENS-toestelletje wordt eerst in bruikleen gegeven om te worden uitgeprobeerd. Blijkt het voor u een doeltreffende pijnbestrijder, dan kan het worden aangekocht.
Snel. Bijvoorbeeld tachyardie: snelle hartslag.
Een veelvoorkomend type epileptische aanval (temporaal epilepsie),waarbij de patiënt doelloze bewegingen uitvoert, een starende blik heeft en gedeeltelijk (of geheel ) het bewustzijn verliest. Vaak gaan aan de aanval bepaalde gevoelens vooraf (zie ook Aura). Dit type aanval werd vroeger ook wel 'petit mal aanval' of 'psychomotorische aanval' genoemd.
Een veelvoorkomend type epileptische aanval (temporaal epilepsie),waarbij de patiënt doelloze bewegingen uitvoert, een starende blik heeft en gedeeltelijk (of geheel ) het bewustzijn verliest. Vaak gaan aan de aanval bepaalde gevoelens vooraf (zie ook Aura). Dit type aanval werd vroeger ook wel 'petit mal aanval' of 'psychomotorische aanval' genoemd.
Bij tetraplegie, of quadriplegie, zijn alle vier de ledematen verminderd krachtig. Het kan gaan om een slappe of een spastische krachtsvermindering.
De thalamus verbindt (en filtert prikkels tussen) het ruggenmerg en de grote hersenen. Het is een schakelstation voor impulsen uit de zintuigen, behalve de reuk. Als je geconcentreerd met iets bezig bent, kan de thalamus ervoor zorgen dat je je minder bewust bent van andere impulsen. Ook reguleert de thalamus de slaap.
De tonus is de spanningstoestand van een spier. De tonus kan abnormaal verhoogd zijn (hypertonie). Dit leidt bijvoorbeeld tot spasmen. De tonus kan abnormaal verlaagd zijn (hypotonie). Dit leidt bijvoorbeeld tot een slappe verlamming waarbij de patiënt een lichaamsdeel niet in een bepaalde stand kan houden.
Bevingen bij het maken van willekeurige bewegingen. Intentietremor: het steeds erger trillen van een ledemaat, naar gelang het doel dichterbij komt.
Visual evoked potentials. Bij dit onderzoek worden de zenuwverbindingen tussen het oog en de hersenen onderzocht. Er wordt een elektrode op het hoofd geplakt en de patiënt krijgt een zwart-wit geblokt bord of lichtflitsen te zien. De patiënt hoeft niets te doen en het onderzoek is niet pijnlijk. Gemeten wordt de tijd die het duurt voor de hersenen reageren op de prikkels.
Duizeligheid.
Het zien, de ogen betreffende.
Het zien, de ogen betreffende.
Wet Maatschappelijke Ondersteuning. Opvolger van WVG = Wet Voorzieningen Gehandicapten.







